
3 results
Integrated efficacy analysis from phase 3 studies of investigational microbiome therapeutic, SER-109, in recurrent Clostridioides difficile infection
-
- Journal:
- Antimicrobial Stewardship & Healthcare Epidemiology / Volume 3 / Issue S2 / June 2023
- Published online by Cambridge University Press:
- 29 September 2023, p. s5
-
- Article
-
- You have access
- Open access
- Export citation
-
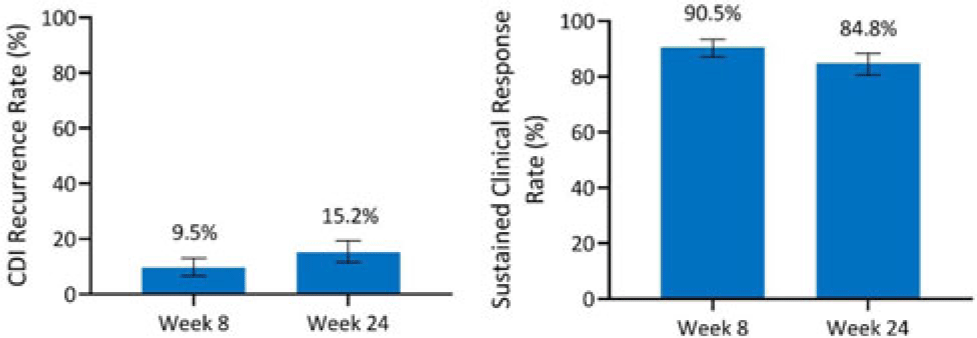
Background: Antibiotics alone are often insufficient to treat recurrent C. difficile infection (rCDI) because they have no activity against C. difficile spores that germinate within a disrupted microbiome. SER-109, an investigational, oral, microbiome therapeutic comprised of purified Firmicutes spores, was designed to reduce rCDI through microbiome repair. We report an integrated efficacy analysis through week 24 for SER-109 from phase 3 studies, ECOSPOR III and ECOSPOR IV. Methods: ECOSPOR III was a randomized, placebo-controlled phase 3 trial conducted at 56 US or Canadian sites that included 182 participants with ≥2 CDI recurrences, confirmed via toxin EIA testing. Participants were stratified by age (<65 years or ≥65 years) and antibiotic regimen (vancomycin, fidaxomicin) and were randomized 1:1 to placebo or SER-109 groups. ECOSPOR IV was an open-label, single-arm study conducted at 72 US or Canadian sites including 263 participants with rCDI enrolled in 2 cohorts: (1) rollover participants from ECOSPOR III who experienced on-study recurrence diagnosed by toxin EIA (n = 29) and (2) participants with ≥1 CDI recurrence (diagnosed by PCR or toxin EIA), inclusive of the current episode (n = 234). In both studies, the investigational product was administered orally as 4 capsules over 3 consecutive days following symptom resolution after standard-of-care antibiotics. The primary efficacy end point was rCDI (recurrent toxin-positive diarrhea requiring treatment) through week 8. Other end points included CDI recurrence rates and safety through 24 weeks. Results: These 349 participants received at least 1 dose of SER-109 in ECOSPOR III or ECOSPOR IV (mean age 64.2; 68.8% female). Overall, 77 participants (22.1%) enrolled with their first CDI recurrence. Four participants received blinded SER-109 in ECOSPOR III followed by a second dose of open-label SER-109 in ECOSPOR IV. Overall, the proportion of participants who received any dose of SER-109 with rCDI at week 8 was 9.5% (33 of 349; 95% CI, 6.6 %–13.0%), and the CDI recurrence rate remained low through 24 weeks (15.2%, 53 of 349; 95% CI, 11.6%–19.4%), corresponding to sustained clinical response rates of 90.5% (95% CI, 87.0%–93.4%) and 84.8% (95% CI, 80.6%–88.4%), respectively (Fig. 1). Most rollover participants (25 of 29, 86.2%) were from the placebo arm; 13.8% had rCDI by week 8. Conclusions: In this integrated analysis, the rates of rCDI were low and durable in participants who received the investigational microbiome therapeutic SER-109, with sustained clinical response rates of 90.5% and 84.8% at weeks 8 and 24, respectively. These data further support the potential benefit of microbiome repair with SER-109 following antibiotics for rCDI to prevent recurrence in high-risk patients.
Financial support: This study was funded by Seres Therapeutics.
Disclosure: None

Integrated safety analysis of phase 3 studies for investigational microbiome therapeutic, SER-109, in recurrent CDI
-
- Journal:
- Antimicrobial Stewardship & Healthcare Epidemiology / Volume 3 / Issue S2 / June 2023
- Published online by Cambridge University Press:
- 29 September 2023, pp. s44-s45
-
- Article
-
- You have access
- Open access
- Export citation
50 Adjunctive Buprenorphine/Samidorphan Combination in Patients with Major Depressive Disorder: Phase 3 Long-term Extension Study Results
-
- Journal:
- CNS Spectrums / Volume 24 / Issue 1 / February 2019
- Published online by Cambridge University Press:
- 12 March 2019, pp. 203-204
-
- Article
-
- You have access
- Export citation
